Plotting
Mixin for convenient plots/visualization of pAnnData object. Typically a wrapper of functions from the scpviz.plotting module.
PlotMixin
Source code in src/scpviz/pAnnData/plot.py
7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476 477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495 496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 512 513 514 515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 544 545 546 547 548 549 550 551 552 553 554 555 556 557 558 559 560 561 562 563 564 565 566 567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 585 586 587 588 589 590 591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 606 607 608 609 610 611 612 613 614 615 616 617 618 619 620 621 622 623 624 625 626 627 628 629 630 631 632 633 634 635 636 637 638 639 640 641 642 643 644 645 646 647 648 649 650 651 652 653 654 655 656 657 658 659 660 661 662 663 664 665 666 667 668 669 670 671 672 673 674 675 676 677 678 679 680 681 682 683 684 685 686 687 688 689 690 691 692 693 694 695 696 697 698 699 700 701 702 703 704 705 706 707 708 709 710 711 712 713 714 715 716 717 718 719 720 721 722 723 724 725 726 727 728 729 730 731 732 733 734 735 736 737 738 739 740 741 742 743 744 745 746 747 748 749 750 751 752 753 754 755 756 757 758 759 760 761 762 763 764 765 766 767 768 769 770 771 772 773 774 775 776 777 778 779 780 781 782 783 784 785 786 787 788 789 790 791 792 793 794 795 796 797 798 799 800 801 802 803 804 805 806 807 808 809 810 811 812 813 814 815 816 817 818 819 820 821 822 823 824 825 826 827 828 829 830 831 832 833 834 835 836 837 838 839 840 841 842 843 844 845 846 847 848 849 850 851 852 853 854 855 856 857 858 859 860 861 862 863 864 865 866 867 868 869 870 871 872 873 874 875 876 877 878 879 880 881 882 883 884 885 886 887 888 889 890 891 892 893 894 895 896 897 898 899 900 901 902 903 904 905 906 907 908 909 910 911 912 913 914 915 916 917 918 919 920 921 922 923 924 925 926 927 928 929 930 931 932 933 934 935 936 937 938 939 940 941 942 943 944 945 946 947 948 949 950 951 952 953 954 955 956 957 958 959 960 961 962 963 964 965 966 967 968 969 970 971 972 973 974 975 976 977 978 979 980 981 982 983 984 985 986 987 988 989 990 991 992 993 994 995 996 997 998 999 1000 1001 1002 1003 1004 1005 1006 1007 1008 1009 1010 1011 1012 1013 1014 1015 1016 1017 1018 1019 1020 1021 1022 1023 1024 1025 1026 1027 1028 1029 1030 1031 1032 1033 1034 1035 1036 1037 1038 1039 1040 1041 1042 1043 1044 1045 1046 1047 1048 1049 1050 1051 1052 1053 1054 1055 1056 1057 1058 1059 1060 1061 1062 | |
plot_abundance
plot_abundance(
ax=None,
namelist=None,
layer="X",
on="protein",
classes=None,
return_df=False,
order=None,
palette=None,
log=True,
facet=None,
height=4,
aspect=0.5,
plot_points=True,
x_label="gene",
kind="auto",
**kwargs: Any
)
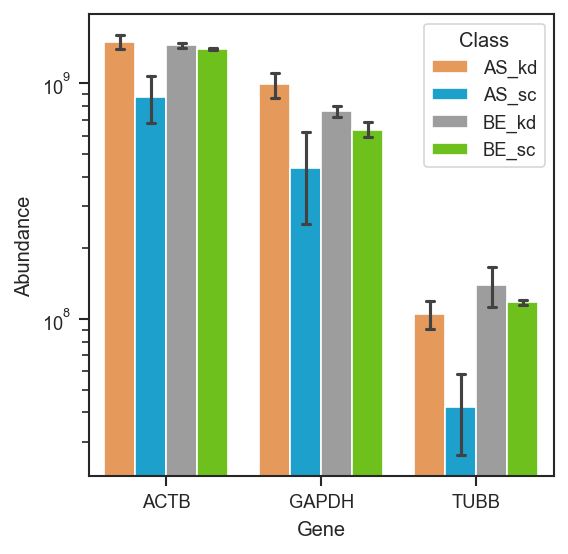
Plot abundance of proteins or peptides across samples.
Thin wrapper around :func:scpviz.plotting.plot_abundance.
This function visualizes expression values for selected proteins or peptides using violin + box + strip plots, or bar plots when the number of replicates per group is small. Supports grouping, faceting, and custom ordering.
Important default behavior:
- This method defaults to log=True (log2-transformed abundances on a linear y-axis).
- With log=False, abundances remain raw and the y-axis is log10-scaled, so the
plot displays log10(abundance) without transforming the underlying values.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ax
|
Axes
|
Axis to plot on. Ignored if |
None
|
namelist
|
list of str
|
List of accessions or gene names to plot. If None, all available features are considered. |
None
|
layer
|
str
|
Data layer to use for abundance values. Default is |
'X'
|
on
|
str
|
Data level to plot, either |
'protein'
|
classes
|
str or list of str
|
|
None
|
return_df
|
bool
|
If True, returns the DataFrame of replicate and summary values. |
False
|
order
|
dict or list
|
Custom order of classes. For dictionary input,
keys are class names and values are the ordered categories.
Example: |
None
|
palette
|
list or dict
|
Color palette mapping groups to colors. |
None
|
log
|
bool
|
If True, apply log2 transformation to abundance values (default here). If False, raw values are used and the y-axis is log10-scaled. |
True
|
facet
|
str
|
|
None
|
height
|
float
|
Height of each facet plot. Default is 4. |
4
|
aspect
|
float
|
Aspect ratio of each facet plot. Default is 0.5. |
0.5
|
plot_points
|
bool
|
Whether to overlay stripplot of individual samples. |
True
|
x_label
|
str
|
Label for the x-axis, either |
'gene'
|
kind
|
str
|
Type of plot. Options:
|
'auto'
|
**kwargs
|
Any
|
Additional keyword arguments passed to seaborn plotting functions. |
{}
|
Returns:
| Name | Type | Description |
|---|---|---|
ax |
Axes or FacetGrid
|
The axis or facet grid containing the plot. |
df |
(DataFrame, optional)
|
Returned if |
Example
Plot abundance of selected marker proteins grouped by cell line and condition:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(4, 4))
pdata.plot_abundance(
ax, namelist=["GAPDH", "TUBB", "ACTB"], classes=["cellline", "condition"]
)
plt.show()
Source code in src/scpviz/pAnnData/plot.py
plot_abundance_boxgrid
plot_abundance_boxgrid(
namelist=None,
ax=None,
layer="X",
on="protein",
classes="Grouping",
return_df=False,
order=None,
plot_type="box",
log_scale=False,
figsize=(2, 2),
palette=None,
y_min=None,
y_max=None,
label_x=True,
show_n=False,
global_legend=True,
box_kwargs=None,
hline_kwargs=None,
bar_kwargs=None,
bar_error="sd",
violin_kwargs=None,
text_kwargs=None,
strip_kwargs=None,
sig_pairs=None,
sig_kwargs=None,
nd_kwargs=None,
)
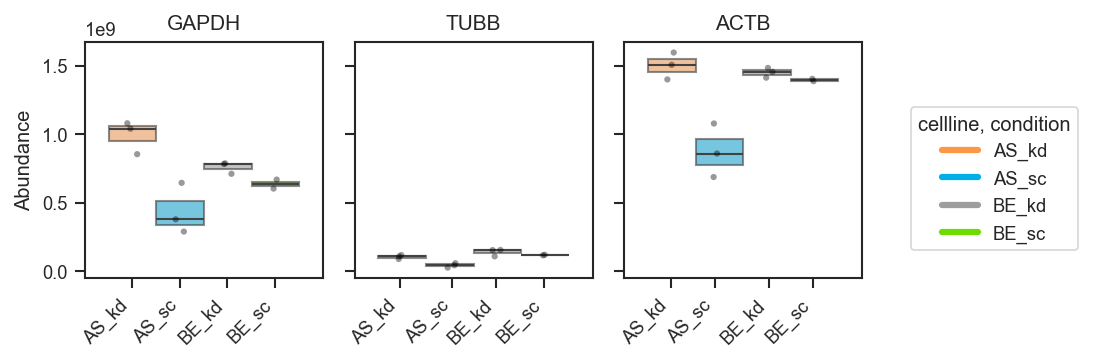
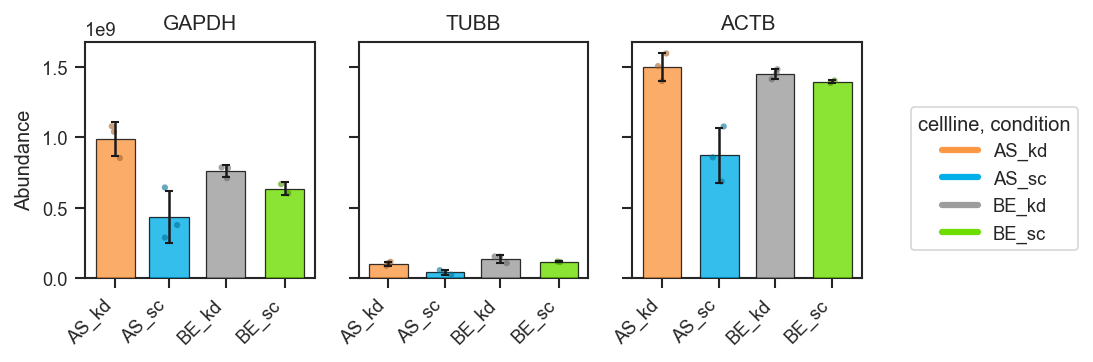
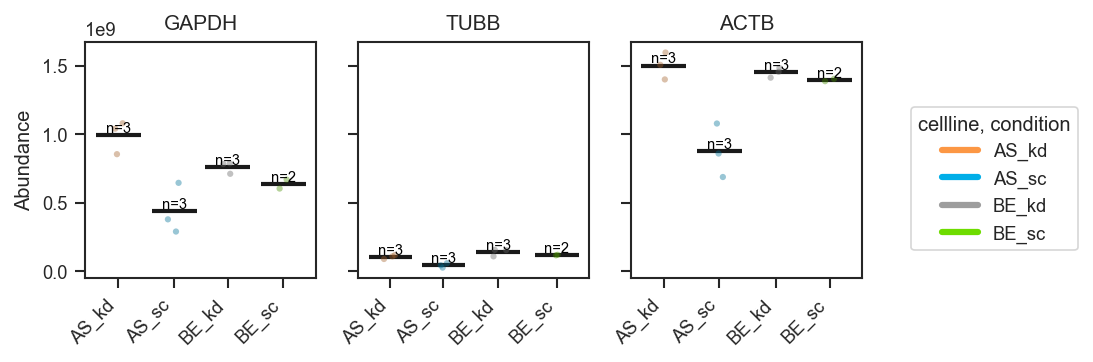
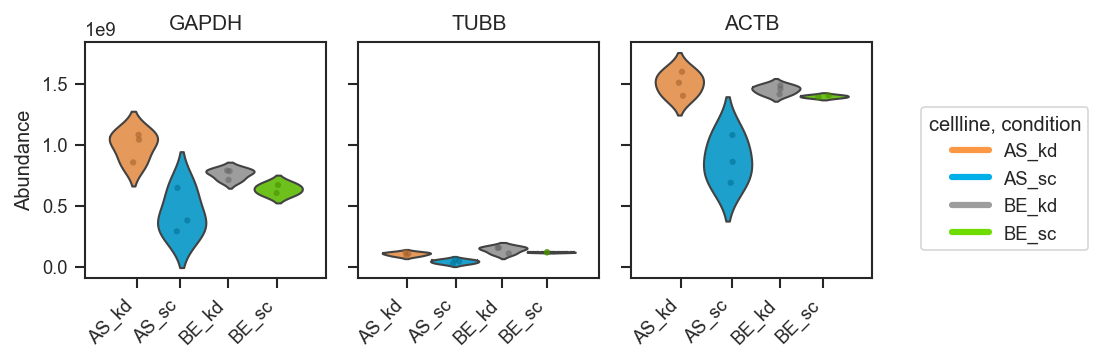
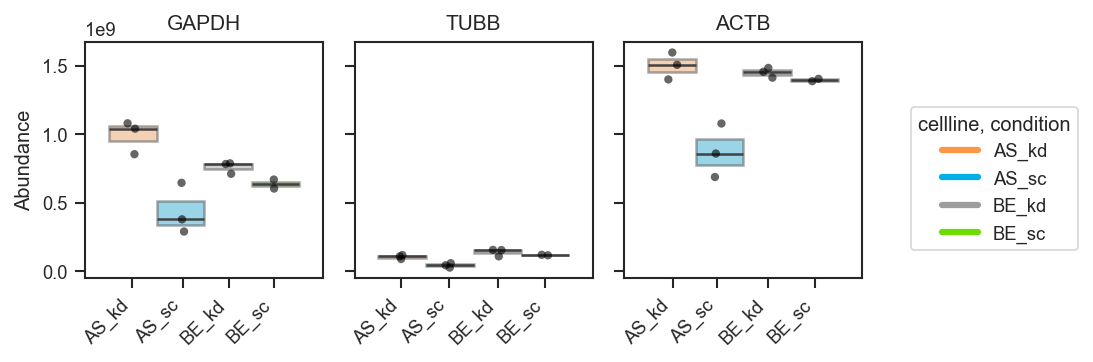
Plot abundance values in a one-row panel of boxplots, mean-lines, bars, or violins.
This function generates a clean horizontal panel, with one subplot per gene,
using plot_type to select boxplots (default), mean-lines, bar plots, or
violin plots. If log_scale=True, abundance values are visualized in
log10 units (with zero or negative values clipped to 0 before transformation).
The layout is optimized for compact manuscript figure panels and supports
custom global legends, count annotations, and flexible formatting via keyword
dictionaries.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
namelist
|
list of str
|
List of accessions or gene names to plot. If None, all available features are considered. |
None
|
ax
|
Axes
|
Axis to plot on. Generates a new axis if None. |
None
|
layer
|
str
|
Data layer to use for abundance values. Default is |
'X'
|
on
|
str
|
Data level to plot, either |
'protein'
|
classes
|
str or list of str
|
Column in |
'Grouping'
|
return_df
|
bool
|
If True, returns the DataFrame of replicate and summary values. |
False
|
order
|
list of str
|
Ordered list to plot by. If None, plots by given dataframe order. |
None
|
plot_type
|
str
|
Type of plot, select from one of |
'box'
|
log_scale
|
bool
|
If True, plot log10-transformed abundances on a linear axis. If False (default), plot raw abundance values on a linear axis. |
False
|
figsize
|
tuple
|
Figure size as (width, height) in inches. |
(2, 2)
|
palette
|
dict or list
|
Color palette for grouping categories.
Defaults to |
None
|
y_min
|
float or None
|
Lower y-axis limit in plotting units. If |
None
|
y_max
|
float or None
|
Upper y-axis limit in plotting units. If |
None
|
label_x
|
bool
|
Whether to display x tick labels inside each subplot. |
True
|
show_n
|
bool
|
Whether to annotate each subplot with sample counts. |
False
|
global_legend
|
bool
|
Whether to display a single global legend. |
True
|
box_kwargs
|
dict
|
Additional arguments passed to |
None
|
hline_kwargs
|
dict
|
Styling for mean segments when |
None
|
bar_kwargs
|
dict
|
Passed to |
None
|
bar_error
|
str
|
Error bar for bar plot. Select from one of
|
'sd'
|
violin_kwargs
|
dict
|
Additional arguments passed to |
None
|
text_kwargs
|
dict
|
Keyword arguments for count labels (e.g., fontsize, offset). |
None
|
strip_kwargs
|
dict
|
Keyword arguments for strip (raw points),
e.g. |
None
|
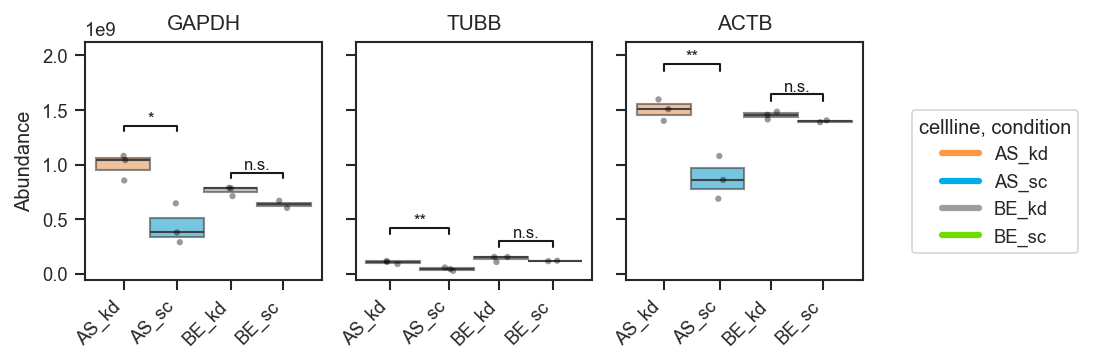
sig_pairs
|
list, bool, or None
|
Pairwise comparisons for significance brackets.
|
None
|
sig_kwargs
|
dict
|
Significance options merged onto defaults
|
None
|
nd_kwargs
|
dict
|
Not-detected annotation options merged onto defaults
|
None
|
Returns:
| Name | Type | Description |
|---|---|---|
fig |
Figure
|
The generated figure. |
axes |
list of matplotlib.axes.Axes
|
One axis per gene. |
df |
(DataFrame, optional)
|
Returned if |
stats_df |
(DataFrame, optional)
|
Returned if |
Note
Default customizations for keyword dictionaries:
Boxplot styling (used when plot_type="box"):
box_kwargs = {
"showcaps": False,
"whiskerprops": {"visible": False},
"showfliers": False,
"boxprops": {"alpha": 0.6, "linewidth": 1},
"linewidth": 1,
"dodge": True,
}
Mean-line styling (used when plot_type="line"):
half_width is in x-axis units; lower it when several classes are dodged
and mean segments would cross.
Bar styling (used when plot_type="bar"):
bar_kwargs = {
"alpha": 0.8,
"edgecolor": "black",
"linewidth": 0.6,
"width": 0.3,
"capsize": 2,
"zorder": 3,
}
width is passed to Axes.bar (x-axis units); use a smaller value when
bars from neighboring hue levels overlap.
Violin styling (used when plot_type="violin"):
Strip styling (raw points; used for all plot types):
Text annotation styling (used when show_n=True):
Example
Basic usage (grouped boxplots):
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="box",
figsize=(2, 2.5),
)
plt.show()
Bar plots with error bars:
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="bar",
bar_error="sd", # "sd", "sem", None, or callable
bar_kwargs={"width": 0.14}, # narrower bars when many groups dodge
figsize=(2, 2.5),
)
plt.show()
Mean-lines with count annotations:
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="line",
show_n=True,
hline_kwargs={"half_width": 0.08}, # shorter segments when groups dodge
figsize=(2, 2.5),
)
plt.show()
Violin plots (distribution-focused):
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="violin",
figsize=(2, 2.5),
)
plt.show()
Customizing appearance (palette, order, and styling):
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="box",
box_kwargs={"boxprops": {"alpha": 0.45}, "linewidth": 1.2},
strip_kwargs={"size": 4, "alpha": 0.6},
figsize=(2, 2.5),
)
plt.show()
Return the plotting DataFrame for downstream checks:
fig, axes, df = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
plot_type="box",
return_df=True,
)
display(df.head())
plt.show()
Significance brackets (explicit pairs, volcano-style dicts):
fig, axes, df, stats = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
sig_pairs=[
({"cellline": "BE", "condition": "sc"}, {"cellline": "BE", "condition": "kd"}),
({"cellline": "AS", "condition": "sc"}, {"cellline": "AS", "condition": "kd"}),
],
sig_kwargs={"fontsize": 8},
return_df=True,
)
plt.show()
Multiple comparisons with a shared group (same group may appear in more than one pair):
fig, axes = pdata.plot_abundance_boxgrid(
namelist=["GAPDH", "TUBB", "ACTB"],
classes=["cellline", "condition"],
sig_pairs=[
({"cellline": "BE", "condition": "sc"}, {"cellline": "BE", "condition": "kd"}),
({"cellline": "BE", "condition": "kd"}, {"cellline": "AS", "condition": "kd"}),
],
sig_kwargs={"fontsize": 8},
)
plt.show()
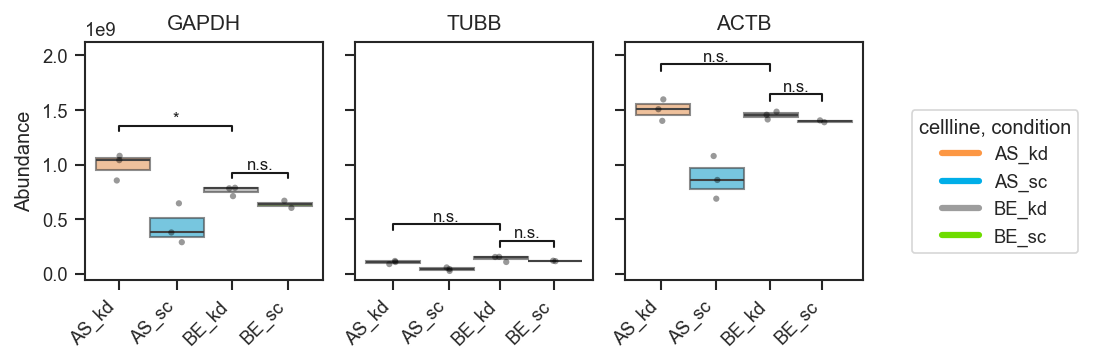
Two hue groups only — auto comparison:
Source code in src/scpviz/pAnnData/plot.py
143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 | |
plot_counts
Violin plot of per-sample count metrics from pdata.summary.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
classes
|
Column in |
None
|
|
y
|
str
|
Column to plot (default |
'protein_count'
|
**kwargs
|
Forwarded to |
{}
|
Source code in src/scpviz/pAnnData/plot.py
plot_pairwise_correlation
plot_pairwise_correlation(
classes: str | list[str],
on: str = "protein",
layer: str = "X",
method: str = "pearson",
order: list | None = None,
show_samples: bool = False,
cmap: str = "RdBu_r",
vmin: float | None = None,
vmax: float | None = None,
annotation_cmap: str | dict | list = "default",
figsize: tuple | None = None,
text_size: int = 9,
colorbar_label: str | None = None,
annot: bool = False,
annot_fmt: str = ".2f",
annot_size: int = 7,
title: str | None = None,
force: bool = False,
subset_mask=None,
show_annotation_legend: bool = True,
legend_anchor_x: float = 0.3,
show_ticklabels: bool | None = None,
ticklabels_auto_max_samples: int = 20,
)
Plot a pairwise protein/peptide abundance correlation heatmap across groups or samples in .obs.
Thin wrapper around :func:scpviz.plotting.plot_pairwise_correlation.
Automatically runs :meth:~scpviz.pAnnData.pAnnData.pairwise_correlation if
results are not already cached (or if force=True). The figure is created
internally; no ax argument is needed.
Cached analysis results are reused when classes, method, layer, and
subset_mask (via the same key as pairwise_correlation) match. If
show_samples=True but the cache lacks a sample matrix, analysis is rerun with
compute_sample_matrix=True. Group-level plots may reuse a cache that already
includes a sample matrix (nothing is stripped). Display order is applied only
when drawing and does not require recomputation.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
classes
|
str | list[str]
|
|
required |
on
|
str
|
|
'protein'
|
layer
|
str
|
Data layer (default |
'X'
|
method
|
str
|
|
'pearson'
|
order
|
list | None
|
Optional row/column order. Must match the matrix being plotted:
If |
None
|
show_samples
|
bool
|
If False (default), plot the group × group matrix. If True,
plot the sample × sample matrix (requires |
False
|
cmap
|
str
|
Matplotlib colormap for the heatmap. |
'RdBu_r'
|
vmin
|
float | None
|
Colormap lower limit; correlation methods default to |
None
|
vmax
|
float | None
|
Colormap upper limit; correlation methods default to |
None
|
annotation_cmap
|
str | dict | list
|
|
'default'
|
figsize
|
tuple | None
|
|
None
|
text_size
|
int
|
Base font size for ticks, colorbar, and legends. |
9
|
colorbar_label
|
str | None
|
Override colorbar label. |
None
|
annot
|
bool
|
If True, write numeric values in each cell. |
False
|
annot_fmt
|
str
|
Format string for cell annotations (e.g. |
'.2f'
|
annot_size
|
int
|
Font size for cell annotations. |
7
|
title
|
str | None
|
Optional figure suptitle. |
None
|
force
|
bool
|
If True, recompute |
False
|
subset_mask
|
Boolean mask or boolean |
None
|
|
show_annotation_legend
|
bool
|
If True (default), draw one legend per annotation track in a dedicated GridSpec column right of the colorbar. |
True
|
legend_anchor_x
|
float
|
Horizontal anchor for annotation legends inside the legend
column, in axes coordinates ( |
0.3
|
show_ticklabels
|
bool | None
|
When |
None
|
ticklabels_auto_max_samples
|
int
|
When |
20
|
Returns:
| Type | Description |
|---|---|
|
|
Note
Heatmap row (y) tick labels are always omitted (symmetric matrix; x-axis labels
carry sample or group names as applicable).
The order argument lists group labels when show_samples=False
(including combined strings such as "AS, kd" for multi-column classes),
but lists observation names when show_samples=True.
Raises:
| Type | Description |
|---|---|
ValueError
|
If |
Example
Group-level heatmap (show_samples=False, default):
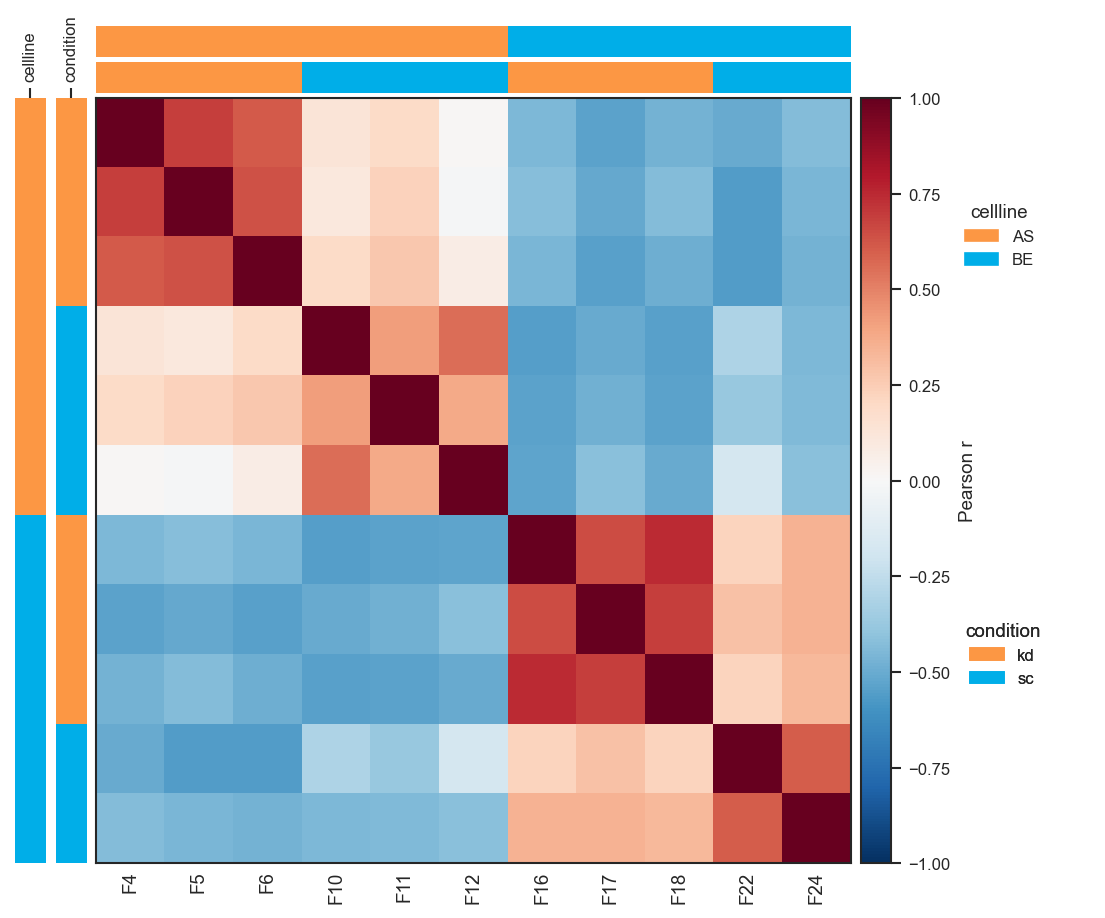
Sample × sample heatmap with x-axis sample names forced on:
fig, ax = pdata.plot_pairwise_correlation(
classes="cellline",
show_samples=True,
show_ticklabels=True,
)
Custom row/column order without recomputing (group labels must match the matrix):
fig, ax = pdata.plot_pairwise_correlation(
classes=["cellline", "treatment"],
order=["AS, kd", "BE, sc", "AS, sc", "BE, kd"],
)
Subset of samples and no annotation legends:
Source code in src/scpviz/pAnnData/plot.py
439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476 477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495 496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 512 513 514 515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 544 545 546 547 548 549 550 551 552 553 554 555 556 557 558 559 560 561 562 563 564 565 566 567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 585 586 587 588 589 590 591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 | |
plot_pca_gsea_bubble
plot_pca_gsea_bubble(
ax,
on="protein",
key_added="pca_gsea",
pcs=None,
top_n=20,
fdr_cutoff=0.1,
size_scale=120.0,
cmap="coolwarm",
title_case_labels=True,
force=False,
gsea_kwargs=None,
top_n_mode="balanced",
include_pathways=None,
exclude_pathways=None,
return_df=False,
)
Plot PCA-GSEA results as a bubble chart (principal component versus pathway).
Thin wrapper around :func:scpviz.plotting.plot_pca_gsea_bubble.
Bubble color encodes NES; bubble area reflects significance (-log10(FDR)). Rows and columns
are ordered by pathway and PC. If pcs is omitted, all PCs present in stored results are used.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ax
|
Axes
|
Target axis. |
required |
on
|
str
|
Data level, |
'protein'
|
key_added
|
str
|
|
'pca_gsea'
|
pcs
|
list of int or None
|
1-based PCs to include; |
None
|
top_n
|
int
|
Cap on distinct pathways after ranking; must be >= 1. |
20
|
fdr_cutoff
|
float or None
|
Same meaning as in |
0.1
|
size_scale
|
float
|
Multiplier for bubble area from |
120.0
|
cmap
|
str or Colormap
|
Colormap for NES-centered coloring. |
'coolwarm'
|
title_case_labels
|
bool
|
If True, format pathway tick labels for display. |
True
|
force
|
bool
|
If True, re-run |
False
|
gsea_kwargs
|
dict or None
|
Forwarded to |
None
|
top_n_mode
|
str
|
|
'balanced'
|
include_pathways
|
str, iterable, or None
|
Keep only pathways matching these names. |
None
|
exclude_pathways
|
str, iterable, or None
|
Remove pathways matching these names. |
None
|
return_df
|
bool
|
If True, return |
False
|
Returns:
| Type | Description |
|---|---|
|
matplotlib.axes.Axes, or |
Example
Bubble chart for the first three PCs, top 25 pathways:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(6, 8))
ax, df = pdata.plot_pca_gsea_bubble(
ax,
pcs=[1, 2, 3],
top_n=25,
return_df=True,
)
Stricter FDR cutoff and title-case pathway labels:
Source code in src/scpviz/pAnnData/plot.py
889 890 891 892 893 894 895 896 897 898 899 900 901 902 903 904 905 906 907 908 909 910 911 912 913 914 915 916 917 918 919 920 921 922 923 924 925 926 927 928 929 930 931 932 933 934 935 936 937 938 939 940 941 942 943 944 945 946 947 948 949 950 951 952 953 954 955 956 957 958 959 960 961 962 963 964 965 966 967 968 969 970 971 972 973 974 975 976 977 978 979 | |
plot_pca_gsea_heatmap
plot_pca_gsea_heatmap(
ax,
on="protein",
key_added="pca_gsea",
pcs=None,
top_n=30,
fdr_cutoff=0.1,
cmap="coolwarm",
title_case_labels=True,
force=False,
gsea_kwargs=None,
top_n_mode="balanced",
include_pathways=None,
exclude_pathways=None,
return_df=False,
)
Plot a pathway-by-principal-component heatmap of PCA-GSEA NES values.
Thin wrapper around :func:scpviz.plotting.plot_pca_gsea_heatmap.
Cell color is NES; optional top_n trimming uses the same FDR-aware scoring as the bubble plot.
Missing PCs in stored results produce NaN columns and a warning.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ax
|
Axes
|
Target axis. |
required |
on
|
str
|
Data level, |
'protein'
|
key_added
|
str
|
|
'pca_gsea'
|
pcs
|
list of int or None
|
1-based PCs as columns; |
None
|
top_n
|
int
|
Maximum pathways to retain after ranking; must be >= 1. |
30
|
fdr_cutoff
|
float or None
|
Same meaning as in |
0.1
|
cmap
|
str or Colormap
|
Heatmap colormap (diverging around zero is typical). |
'coolwarm'
|
title_case_labels
|
bool
|
If True, format pathway labels on the axis. |
True
|
force
|
bool
|
If True, re-run |
False
|
gsea_kwargs
|
dict or None
|
Forwarded to |
None
|
top_n_mode
|
str
|
|
'balanced'
|
include_pathways
|
str, iterable, or None
|
Keep only pathways matching these names. |
None
|
exclude_pathways
|
str, iterable, or None
|
Remove pathways matching these names. |
None
|
return_df
|
bool
|
If True, return |
False
|
Returns:
| Type | Description |
|---|---|
|
matplotlib.axes.Axes, or |
Example
Heatmap of NES for four PCs and the 40 top-ranked pathways:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(5, 10))
pdata.plot_pca_gsea_heatmap(ax, pcs=[1, 2, 3, 4], top_n=40)
Diverging colormap with formatted pathway names on rows:
Source code in src/scpviz/pAnnData/plot.py
981 982 983 984 985 986 987 988 989 990 991 992 993 994 995 996 997 998 999 1000 1001 1002 1003 1004 1005 1006 1007 1008 1009 1010 1011 1012 1013 1014 1015 1016 1017 1018 1019 1020 1021 1022 1023 1024 1025 1026 1027 1028 1029 1030 1031 1032 1033 1034 1035 1036 1037 1038 1039 1040 1041 1042 1043 1044 1045 1046 1047 1048 1049 1050 1051 1052 1053 1054 1055 1056 1057 1058 1059 1060 1061 1062 | |
plot_pca_gsea_pathway_vectors
plot_pca_gsea_pathway_vectors(
ax,
on="protein",
key_added="pca_gsea",
plot_pc=[1, 2],
n_vectors=plotting.N_VECTORS_UNSET,
fdr_cutoff=0.1,
arrow_scale=0.25,
pca_kwargs=None,
show_samples=True,
title_case_labels=True,
force=False,
gsea_kwargs=None,
adjust_labels=True,
adjust_text_kwargs=None,
text_positions=None,
lock_text_positions=False,
top_n_mode="balanced",
exclude_pathways=None,
namelist=None,
cmap=None,
xlim=None,
ylim=None,
return_df=False,
)
Overlay PCA-GSEA pathways as arrows in a two-dimensional PCA sample space.
Thin wrapper around :func:scpviz.plotting.plot_pca_gsea_pathway_vectors.
Each arrow encodes normalized enrichment scores (NES) on two principal components taken from
adata.uns[key_added]['results'] (from pca_gsea). Arrow endpoints are rescaled using the
current axis limits so pathways remain visible; they are not plotted in the same numeric units as
sample coordinates. When show_samples is True, the sample PCA scatter is drawn first via
plot_pca.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ax
|
Axes
|
Target axis (2D). |
required |
on
|
str
|
Data level, |
'protein'
|
key_added
|
str
|
|
'pca_gsea'
|
plot_pc
|
list of int
|
Exactly two 1-based PCs, e.g. |
[1, 2]
|
n_vectors
|
int, sequence, ``None``, or unset
|
Caps auto-selected pathways (after |
N_VECTORS_UNSET
|
fdr_cutoff
|
float or None
|
For auto-selected rows: pathway-level FDR filtering and score gating.
Namelist pathways skip the row FDR filter; a warning is printed per named pathway when
|
0.1
|
arrow_scale
|
float
|
Scale factor for arrow length relative to axis span. |
0.25
|
pca_kwargs
|
dict or None
|
Additional arguments passed to |
None
|
show_samples
|
bool
|
If True, plot samples first; if False, draw only axes, grid lines, and arrows. |
True
|
title_case_labels
|
bool
|
If True, format pathway labels for display (e.g. title case). |
True
|
force
|
bool
|
If True, re-run |
False
|
gsea_kwargs
|
dict or None
|
Forwarded to |
None
|
adjust_labels
|
bool
|
If True, run |
True
|
adjust_text_kwargs
|
dict or None
|
Extra keyword arguments for |
None
|
text_positions
|
dict or None
|
Optional manual label positions; keys are pathway raw or display
strings, values are |
None
|
lock_text_positions
|
bool
|
If True, labels with entries in |
False
|
top_n_mode
|
str
|
|
'balanced'
|
exclude_pathways
|
str, iterable, or None
|
Remove pathways matching these names (raw Term, short pathway, or library). |
None
|
namelist
|
list of str or None
|
Pathways to always include first. Shown even if they fail FDR;
|
None
|
cmap
|
dict or None
|
Per-pathway colors; lookup raw |
None
|
xlim
|
tuple or None
|
Applied after scatter / empty axes, before arrow scaling. |
None
|
ylim
|
tuple or None
|
Same as |
None
|
return_df
|
bool
|
If True, also return a DataFrame with NES, FDR, and label positions. |
False
|
Returns:
| Type | Description |
|---|---|
|
matplotlib.axes.Axes, or |
Example
Default overlay on PC1 vs PC2 with label de-cluttering:
import matplotlib.pyplot as plt
fig, ax = plt.subplots()
ax, vec_df = pdata.plot_pca_gsea_pathway_vectors(
ax,
plot_pc=[1, 2],
adjust_text_kwargs={"expand": (1.3, 1.3)},
return_df=True,
)
Reuse label positions from a previous run:
Source code in src/scpviz/pAnnData/plot.py
607 608 609 610 611 612 613 614 615 616 617 618 619 620 621 622 623 624 625 626 627 628 629 630 631 632 633 634 635 636 637 638 639 640 641 642 643 644 645 646 647 648 649 650 651 652 653 654 655 656 657 658 659 660 661 662 663 664 665 666 667 668 669 670 671 672 673 674 675 676 677 678 679 680 681 682 683 684 685 686 687 688 689 690 691 692 693 694 695 696 697 698 699 700 701 702 703 704 705 706 707 708 709 710 711 712 713 714 715 716 717 718 719 720 721 722 723 724 725 726 727 728 729 730 731 732 733 734 | |
plot_pca_protein_vectors
plot_pca_protein_vectors(
ax,
on="protein",
plot_pc=(1, 2),
gene_col="Genes",
n_vectors=plotting.N_VECTORS_UNSET,
arrow_scale=0.25,
pca_kwargs=None,
show_samples=True,
title_case_labels=False,
adjust_labels=True,
adjust_text_kwargs=None,
text_positions=None,
lock_text_positions=False,
min_abs_loading_for_top_n=None,
top_n_mode="balanced",
exclude_genes=None,
namelist=None,
cmap=None,
xlim=None,
ylim=None,
return_df=False,
)
Overlay protein PCA loadings as arrows in a two-dimensional sample PCA space.
Thin wrapper around :func:scpviz.plotting.plot_pca_protein_vectors.
Arrows use feature loadings from adata.uns['pca']['PCs'] (from pAnnData.pca), not GSEA NES.
Geometry matches plot_pca_gsea_pathway_vectors: each arrow runs from the origin in the direction
(loading_on_PCx, loading_on_PCy), with length rescaled from the current axis limits for visibility.
Labels default to the gene_col column in .var when present, otherwise .var_names.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ax
|
Axes
|
Target axis (2D). |
required |
on
|
str
|
Data level, |
'protein'
|
plot_pc
|
tuple or list of int
|
Exactly two 1-based PCs. |
(1, 2)
|
gene_col
|
str
|
Column in |
'Genes'
|
n_vectors
|
int, sequence, ``None``, or unset
|
Caps auto-selected proteins (rows not already taken
by |
N_VECTORS_UNSET
|
arrow_scale
|
float
|
Scale factor for arrow length relative to axis span. |
0.25
|
pca_kwargs
|
dict or None
|
Forwarded to |
None
|
show_samples
|
bool
|
If True, draw the sample PCA scatter first; if False, only axes and arrows. |
True
|
title_case_labels
|
bool
|
If True, lightly format gene text (underscores to spaces, title case). |
False
|
adjust_labels
|
bool
|
If True, run |
True
|
adjust_text_kwargs
|
dict or None
|
Extra keyword arguments for |
None
|
text_positions
|
dict or None
|
Manual label positions keyed by gene or formatted label. |
None
|
lock_text_positions
|
bool
|
If True, manual positions are excluded from |
False
|
min_abs_loading_for_top_n
|
float or None
|
If set, ranking scores on a PC are zero when
|
None
|
top_n_mode
|
str
|
|
'balanced'
|
exclude_genes
|
str, iterable, or None
|
Remove genes/features matching these strings (gene label or
|
None
|
namelist
|
list of str or None
|
Gene labels (matrix row index, exact |
None
|
cmap
|
dict or None
|
Map gene label to a matplotlib color; lookup tries raw name, formatted label,
then case-insensitive keys. Default |
None
|
xlim
|
tuple or None
|
Applied after the PCA scatter and before arrow length scaling. |
None
|
ylim
|
tuple or None
|
Same stage as |
None
|
return_df
|
bool
|
If True, return |
False
|
Returns:
| Type | Description |
|---|---|
|
matplotlib.axes.Axes, or |
Example
Show top protein loadings on PC1 vs PC2 on sample PCA scatter:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(4, 4))
pdata.pca(on="protein")
pdata.plot_pca_protein_vectors(ax, n_vectors=10)
plt.show()
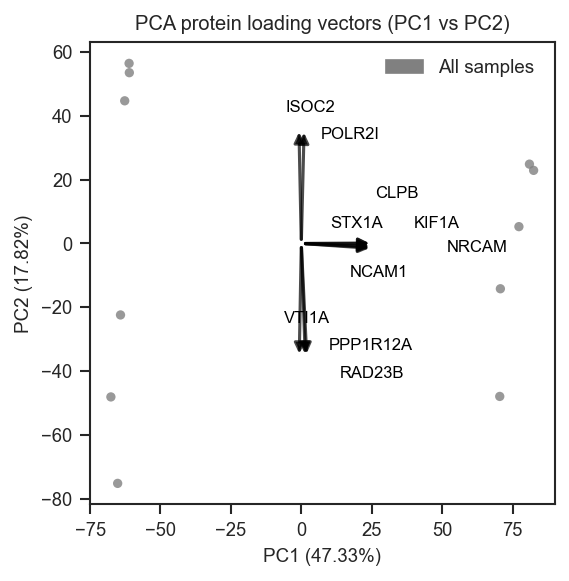
Top-loading genes with returned coordinates:
fig, ax = plt.subplots()
ax, vec = pdata.plot_pca_protein_vectors(
ax,
plot_pc=[1, 2],
n_vectors=25,
return_df=True,
)
Split-axis selection on PC1 and PC3:
fig, ax = plt.subplots()
pdata.plot_pca_protein_vectors(
ax,
plot_pc=[1, 3],
n_vectors=[5, 3],
adjust_labels=False,
)
Explicit genes with colors and axis limits:
fig, ax = plt.subplots()
pdata.plot_pca_protein_vectors(
ax,
plot_pc=[1, 2],
namelist=["TP53", "EGFR"],
cmap={"TP53": "crimson", "egfr": "steelblue"},
xlim=(-6, 6),
ylim=(-5, 5),
)
Loading arrows only (no sample points):
Source code in src/scpviz/pAnnData/plot.py
736 737 738 739 740 741 742 743 744 745 746 747 748 749 750 751 752 753 754 755 756 757 758 759 760 761 762 763 764 765 766 767 768 769 770 771 772 773 774 775 776 777 778 779 780 781 782 783 784 785 786 787 788 789 790 791 792 793 794 795 796 797 798 799 800 801 802 803 804 805 806 807 808 809 810 811 812 813 814 815 816 817 818 819 820 821 822 823 824 825 826 827 828 829 830 831 832 833 834 835 836 837 838 839 840 841 842 843 844 845 846 847 848 849 850 851 852 853 854 855 856 857 858 859 860 861 862 863 864 865 866 867 868 869 870 871 872 873 874 875 876 877 878 879 880 881 882 883 884 885 886 887 | |
plot_rs
Visualize connectivity in the RS (protein × peptide) matrix.
Generates side-by-side histograms:
- Left: Number of peptides mapped to each protein
- Right: Number of proteins associated with each peptide
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
figsize
|
tuple
|
Size of the matplotlib figure (default: (10, 4)). |
(10, 4)
|
Returns:
| Name | Type | Description |
|---|---|---|
out |
None
|
No return value; shows the figure interactively or closes it when using a non-interactive backend. |